Calculation of double differential cross sections for inner-shell excitations in the local density approximation
- Abstract number
- 181
- Event
- European Microscopy Congress 2020
- DOI
- 10.22443/rms.emc2020.181
- Corresponding Email
- [email protected]
- Session
- PST.4 - Spectroscopies in Electron, X-ray and Ion Microscopy
- Authors
- Leonhard Segger (1), Prof. Dr. Helmut Kohl (1)
- Affiliations
-
1. Westfälische Wilhelms-Universität Münster
- Keywords
cross-sections EELS excitations GOS spectra
- Abstract text
Summary
We present a program to calculate double differential cross sections, which can be used to determine chemical compositions from EEL spectra. For the computation of the relevant matrix elements we use wave functions obtained with the local density approximation.
Introduction
To determine the concentrations of the elements in a sample quantitatively, one needs cross sections for the ionization of an electron from a chosen shell losing a specific amount of energy during the interaction and being scattered into a specific solid angle element. Such cross sections as a function of the energy of the incident electrons are currently determined from tabulated values of the generalized oscillator strengths (GOS) obtained using Herman-Skillman wave functions [1]. We show that in modern PCs computing times are short enough to permit the double differential cross section to be calculated ab initio at the time of spectrum analysis.
To compare our results in the local density approximation with the tabulated Herman-Skillman data, we have additionally computed the GOS.Methods
Following the procedures described in [1], we start by computing the wave functions of the initial state. Rather than using Herman-Skillman wave functions, we calculate them self-consistently within the local density approximation using the exchange correlation potential after Perdew [2].
For this the atomic potential is given by.
The summands are respectively the potential from the atomic core, the Coulomb potential given by the electron-electron interaction, and the exchange correlation potential. The exchange correlation potential is then calculated in the local density approximation. With this the potential is iteratively corrected until it converges.
We use a Fortran program by Krüger [3], which is based on a procedure by Hamann [4], whereas the broadly used tabulations by [1] use a Hartree-Slater central field model and Herman-Skillman wave functions for the bound states, which is expected to be less accurate [2].Using this atomic potential the wave function of the ejected free electron is calculated. To normalize the free wave functions they are matched to spherical Bessel and Neumann functions at large distances from the core [5].
The remaining integral constitutes a spherical Bessel transform. Using the convolution theorem this integral is solved rather efficiently with the fast Fourier transformation routine [6].
Results and Discussion
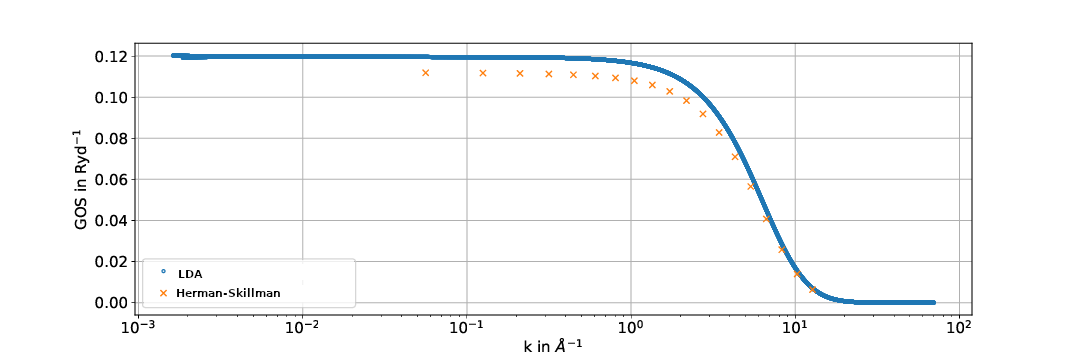
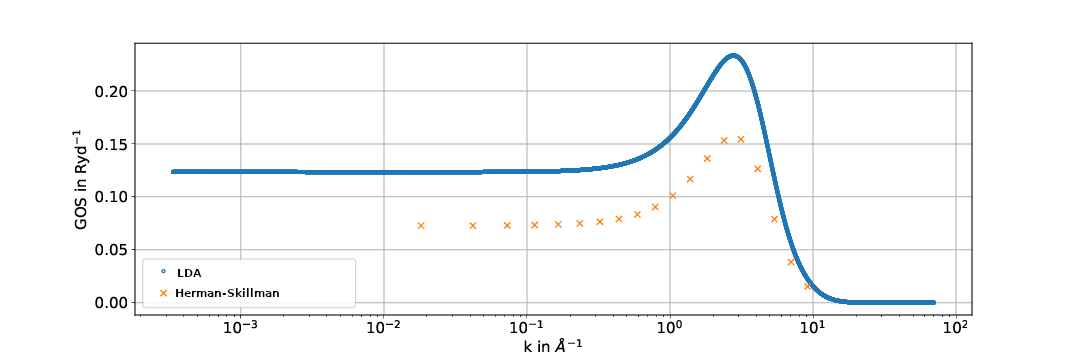
We compare the GOSs calculated in the LDA with the calculations performed previously by [1], where Herman-Skillman wave functions were used as described above.
Generally the results exhibit similar shapes but sometimes differ significantly. This could possibly be attributed to the approximations used in the calculation of the Hartree-Slater central field model being less accurate than the local density approximation used here. To find out whether this is the case comparisons to experimental data are yet to be done.
Fig. 1: Generalized oszillator strengths for the C-K shell. The energy of the ejected electrons is 10.5 eV.
Fig. 2: Generalized oszillator strengths for the Cu-M23 shell. The energy of the ejected electrons is 12.3 eV.
For a specific shell and ten values for the energy of the free electron this calculation takes about 2 seconds for heavy elements (Pb) on a modern PC (Intel Core i3-4150 CPU @ 3.50GHz × 4). For quantification of experimental data this requires computation times of likely less than a minute (multiple shells, multiple energy loss values). Therefore it seems that a tabulation of GOSs is no longer necessary.
Conclusion
We have developed a program to calculate generalized oscillator strengths and double differential cross sections for the quantitative determination of chemical compositions from EEL Spectra. The computations are done within a few seconds thus making a tabulation of the GOSs for subsequent use unnecessary.
Comparing our results with those of previous calculations we observe some differences, particularly for higher shells. To determine, whether the local density approximation is more accurate, we plan to compare the data with experimantal spectra.
Our approach neglects spin effects beyond the computation of the atomic wave functions and potential and does not include relativistic effects. These become important for acceleration voltages of 200 keV and beyond [5].- References
[1] R. D. Leapman, P. Rez and F. Mayers, J. Chem. Phys 72 (1980), p. 1232
[2] J. P. Perdew and A. Zunger, Phys. Rev. B 23 (1981), p. 5048
[3] P. Krüger, personal communication
[4] D. R. Hamann, Phys. Rev. B 40 (1989), p. 2980
[5] M. Frigge, diploma thesis, WWU Münster (2011)
[6] J. D. Talman, Comp. Phys. Comm. 180 (2009), p. 332